| O + N2 | reac 2 |
| N +O2 | reac 3 |
| N + OH | reac 4 |
| O + N2 | reac 2 |
| N +O2 | reac 3 |
| N + OH | reac 4 |
Nitrogen-free fuels thus provide the best means to study the effect of sulphur on the formation of thermal-NOX as this would be formed from fixation of atmospheric nitrogen only. One such example is the experimental work of Wendt and Ekmann (Wendt and Ekmann (1975)) . They observed notable decreases of thermal-NO emissions from methane/air flames at all equivalence ratios, which included fuel-rich and fuel-lean, when SO2 and H2S were added in relatively large amounts (4.9 % SO2 by volume in the fuel). Maximum reductions of 36 % of NO were obtained at slightly sub-stoichiometric conditions. The authors postulated that SO2 was responsible for the inhibition of NO formation, and that H2S acted by being converted to SO2 before interacting with the NO formation process. In that respect, pre-heating of the combustion air enhances NO inhibition by hydrogen sulphide as it accelerates its conversion to SO2, especially in fuel-lean conditions.
More complex fuels were used in the recent work by Hampartsoumian and Nimmo (Hampartsoumian and Nimmo (1995)) who reported reductions of thermal-NO. Several gas oils and fuel oils were doped with tetrahydrothiophene or SO2 (gas) and burned in a staged flame. Although reductions of up to 20 ppmv were caused by sulphur dioxide, such an effect decreased or disappeared when conditions in the initial stages of the flame were relatively fuel-rich (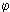 > 1.21). In this study air staging turned out to be the most important single parameter influencing the effect of S on NOX emissions, which may show to have practical implications for NOX reduction strategies in staged burners.
Although the formation of thermal-NO is understood to depend mainly on flame temperature, the reduction observed was explained in terms of the catalytic recombination of radicals by means of third-body reactions such as (Tseregounis and Smith (1984), Zachariah and Smith (1987)) :
> 1.21). In this study air staging turned out to be the most important single parameter influencing the effect of S on NOX emissions, which may show to have practical implications for NOX reduction strategies in staged burners.
Although the formation of thermal-NO is understood to depend mainly on flame temperature, the reduction observed was explained in terms of the catalytic recombination of radicals by means of third-body reactions such as (Tseregounis and Smith (1984), Zachariah and Smith (1987)) :
| X + SO2 + M | reac 60 |
| Y + XSO2 | reac 61 |
where X, Y = O, OH, H Catalytic recombination would in turn reduce the rate of the Zeldovich mechanism. The radical recombination of OH and H radicals by SO2 in fuel-rich combustion conditions had been investigated previously by Durie et al. (Durie et al. (1971)) and Kallend (Kallend (1972)). The mechanism was thought to occur through the reactions:
| H + SO2 + M | reac 62 |
| H + HSO2 | reac 63 |
| OH + HSO2 | reac 64 |
The mechanism is completed with the following slower, uncatalysed recombination reactions:
H + H + M  H2 + M H2 + M
| reac 65 |
| H + OH + M H2O + M
| reac 66 |
| H + H2O | reac -7 |
where reactions 62 and 63 involve H radicals and can modify the pool of radicals established by reactions (Burgess and Langley (1991)):
| O + H2 | reac 5 |
| H + O2 | reac 6 |
| H2 + OH | reac 7 |
| H2O + O OH + OH
| reac 34 |
Kallend (Kallend (1972)) found experimental evidence that SO2 accelerates the recombination of H radicals, decreasing its concentrations shortly after the flame reaction zone. The existence of H radicals can modify the concentrations of reduced sulphur species, such as HS, H2S, S, S2 and SO (Durie et al. (1971)) , with effects on other reactions.
According to the radical recombination scheme (reactions 62, 63 and 64), the HSO2 radical plays a key factor in the process. Numerical simulation was performed by Zachariah and Smith (Zachariah and Smith (1987)) in order to investigate the matter. Although not detected experimentally, their calculations confirmed that the HSO2 radical is essential as an intermediate step in the formation of other radical species. Further work by Halstead and Jenkins (Halstead and Jenkins (1969)) emphasised the relevance of HSO2 radicals in the catalytic recombination of H radicals by SO2 in H2/O2/N2 flames. They concluded that the recombination of H radicals is a second order process which is rate-controlled by reactions 63 and 64:
| H + HSO2 | reac 63 |
| OH + HSO2 | reac 64 |
However, Kallend (Kallend (1972)) remarked that another effect of addition of SO2 is the change of adiabatic flame temperature. Experimental results in rich H2/O2/N2 flames revealed that SO2 was able to rise the flame temperature in fuel-rich flames, with temperature increases of up to 200 °C in the most extreme cases. This change of temperature determined that the distribution of radicals and S species varied according to stoichiometry and amount of sulphur added.
One of the most comprehensive studies on N-S interactions published to date is that of Pfefferle and Churchill (Pfefferle and Churchill (1989)) which included both experimental results and numerical modelling of the effect of fuel-S on nitrogenous species. The experimental work was performed in a thermally stabilised burner (TSB), where combustion of ethane, air, ammonia (fuel-N) and hydrogen sulphide (fuel-S) was forced past a hot surface. The numerical model comprised a variety of sulphur reactions (oxidation, reactions of SO2 and SO2 with atomic oxygen, sulphur-nitrogen and sulphur-carbon) in a simulated TSB burner (the so-called "sparse" model). In their experimental results thermal-NOX was reduced on addition of sulphur. Almost no difference in the small decrease of thermal-NOX (between 5 and 10 %) was observed with different levels of S doping. The decrease of thermal-NOX was thought to be, again, due to the decrease of the concentrations of H and OH radicals, which in turn reduced the rates of reactions 2, 3 and 4 of the Zeldovich mechanism.
Nitrogen-sulphur interactions discussed so far are based on indirect routes through intermediate species that would inhibit the formation of NO. In addition, nitric oxide can also be recombined after its formation. Direct nitrogen-sulphur post-formation reactions were postulated by Chagger et al. (Chagger et al. (1991)) to explain small decreases of nitric oxide in the slightly fuel-rich conditions of methane flames with re-burning (the re-burn fuel was also methane):
| SH + NO NS + OH
| reac 67 |
| SH + NO SO + NH
| reac 68 |
| SH + NO S + HNO
| reac 69 |
where SH would be formed by the reduction of SO2 via H2S in rich conditions. The latter reaction 69 is unlikely to constitute a major NO reduction path as the formation of HNO would lead to NO regeneration.
Chagger et al. (Chagger et al. (1991)) deemed that the most likely path for nitrogen recombination was reaction 67, followed by other reactions as in the following scheme:
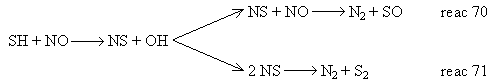
However, the overall process turned out to be inefficient for NOX reduction as 0.1 % SO2 caused only a small further 1-2 % NOX reduction, whereas the sole addition of methane re-burning achieved reductions up to 65 % over the initial NOX emissions.
Other sulphur species can also interact with NOX after its formation. Lyon et al. (Lyon et al. (1990)) reported decreases in the conversion of NO into NO2 by methanol when SO2 was present in the low-temperature (700 - 800 °C) post-combustion gases of coal or natural gas burning. SO2 was concurrently reduced to SO2 as both NO and SO2 competed for the HO2 radicals supplied by the combustion of methanol:
| NO + HO2 | reac 29 |
| SO2 + HO2 | reac 72 |
| HSO2 + M SO2 + OH + M
| reac 73 |
A further similar study was carried out by Zamansky et al. (Zamansky et al. (1996)) , in which hydrogen peroxide was the source of HO2 radicals. Removal efficiencies of up to 98 and 85 % were achieved for NO and SO2, respectively.
The efficient displacement of nitrogen species towards NO2 could have practical implications for NOX removal, as SO2 scrubbers are also capable of eliminating NO2, but not nitric oxide.
 Previous |  Table of Contents |  Next |